La règle des 80-125 % : comprendre les intervalles de confiance en bioéquivalence
 juil., 16 2026
juil., 16 2026
Vous avez probablement déjà entendu dire que les médicaments génériques peuvent contenir jusqu'à 25 % de principe actif en moins que leurs équivalents de marque. C'est une idée reçue tenace qui circule dans les couloirs des pharmacies et sur les forums patients. La réalité est bien plus nuancée, et elle repose sur un concept statistique précis : la règle des 80-125 %, définie comme un critère réglementaire international utilisé pour établir la bioéquivalence entre deux formulations pharmaceutiques. Ce n'est pas une tolérance sur le poids de la pilule, mais une mesure de la façon dont votre corps absorbe le médicament.
Pour comprendre pourquoi cette fourchette spécifique existe, il faut remonter à la fin des années 1970. À l'époque, la FDA (Food and Drug Administration) américaine cherchait un moyen d'autoriser les génériques sans refaire des essais cliniques coûteux et longs pour chaque nouvelle version. L'objectif était simple : prouver que le générique se comporte de la même manière dans le sang que le médicament de référence. Le résultat a été l'adoption d'un standard mondial qui garantit que la différence d'absorption reste cliniquement insignifiante pour la grande majorité des patients.
Que mesurent vraiment ces 80-125 % ?
Le cœur du sujet réside dans deux paramètres pharmacocinétiques clés : l'AUC (Area Under the Curve, ou surface sous la courbe de concentration plasmatique) et le Cmax (concentration maximale). L'AUC représente l'exposition totale du corps au médicament sur une période donnée, tandis que le Cmax indique la vitesse à laquelle le médicament atteint son pic dans le sang.
Lorsqu'on compare un générique à un original, on ne regarde pas si chaque patient absorbe exactement la même quantité. On utilise plutôt une moyenne géométrique. La règle exige que l'intervalle de confiance à 90 % du rapport entre ces moyennes tombe entièrement entre 80 % et 125 %. Concrètement, cela signifie qu'on peut affirmer avec 90 % de certitude statistique que la différence d'absorption réelle est inférieure à 20 % dans chaque sens.
Pourquoi 90 % et non 95 %, comme on le voit souvent en statistiques classiques ? Parce qu'il s'agit d'un test bilatéral. On accepte un risque d'erreur de 5 % à la borne inférieure et de 5 % à la borne supérieure, ce qui fait un risque total de 10 %. Cette méthode assure que ni le générique ne soit trop peu absorbé (moins de 80 %), ni trop rapidement (plus de 125 %).
La transformation logarithmique : la clé mathématique
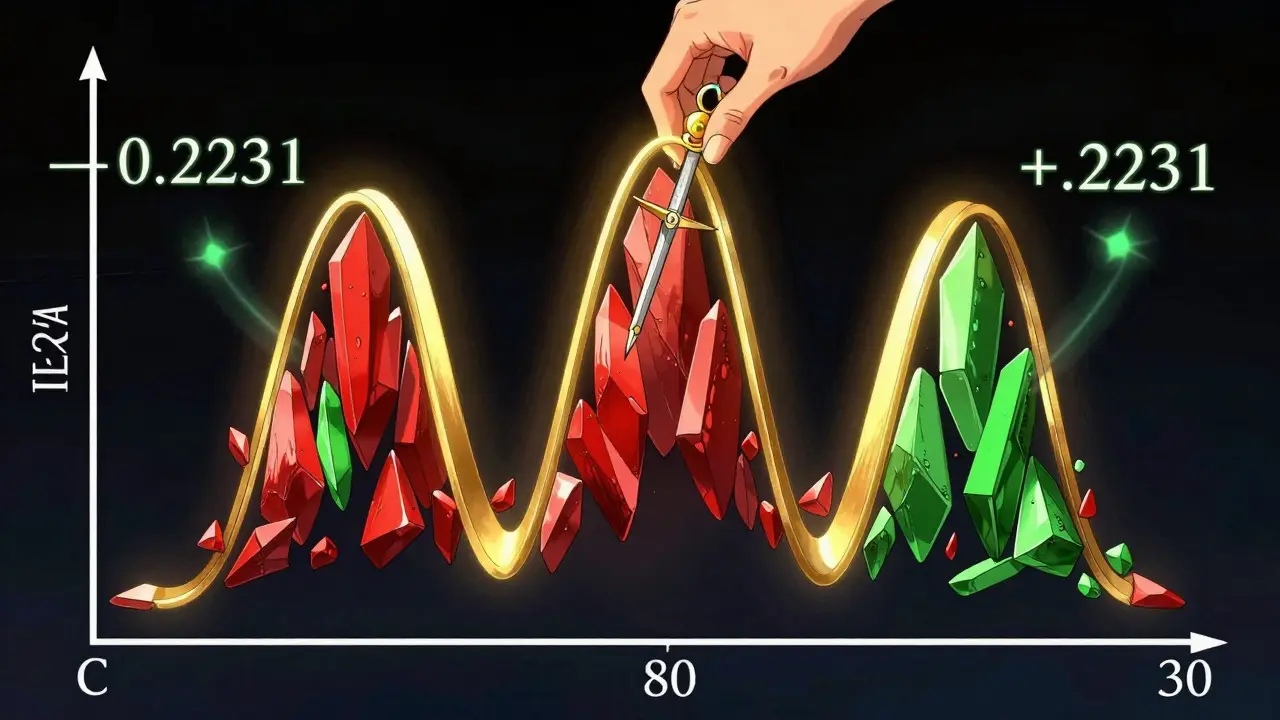
Il y a une subtilité technique cruciale que beaucoup ignorent : les données pharmacocinétiques ne suivent pas une distribution normale classique. Elles sont asymétriques. Pour pouvoir appliquer des tests statistiques valides, les chercheurs transforment les données AUC et Cmax en utilisant des logarithmes.
Sur cette échelle logarithmique, l'intervalle de 80-125 % devient symétrique autour de zéro. Les bornes correspondent à -0,2231 et +0,2231. Cette symétrie est essentielle car elle traite les écarts positifs et négatifs de manière équitable. Sans cette transformation, une différence de 20 % vers le bas n'aurait pas la même pondération statistique qu'une différence de 20 % vers le haut, faussant ainsi l'analyse.
Ce processus explique pourquoi on parle de "rapport des moyennes géométriques". Une fois l'analyse terminée, on retransforme les résultats pour obtenir les pourcentages familiers de 80 à 125 %. C'est cette rigueur mathématique qui permet aux agences de santé de valider des milliers de génériques chaque année avec une sécurité accrue.
Démystifier les mythes sur les principes actifs
Un malentendu persistant concerne la composition physique des comprimés. Beaucoup pensent que si la règle est de 80-125 %, alors un générique pourrait légalement contenir seulement 80 % de la dose prescrite. C'est faux. La quantité de principe actif dans le comprimé doit être extrêmement précise, généralement entre 95 % et 105 % de la dose étiquetée, tout comme pour les médicaments de marque.
La règle des 80-125 % ne s'applique pas au poids du médicament, mais à sa biodisponibilité. Deux comprimés peuvent avoir exactement la même quantité de molécule active, mais différer par leur excipient (liants, colorants, agents de dissolution). Ces différences peuvent faire varier la vitesse ou l'étendue de l'absorption par l'organisme. C'est cette variation biologique que la règle encadre, pas la formulation chimique brute.
| Critère | Médicament de Marque | Générique Bioéquivalent |
|---|---|---|
| Quantité de principe actif | 95-105 % de la dose nominale | 95-105 % de la dose nominale |
| Intervalle de confiance (AUC/Cmax) | N/A (Référence) | Doit être inclus dans 80-125 % |
| Type d'essai requis | Essais cliniques d'efficacité complets | Étude de bioéquivalence (sujets sains) |
| Coût de développement | Milliards de dollars | 2 à 5 millions de dollars |
Exceptions et cas particuliers
Si la règle des 80-125 % est la norme, elle n'est pas absolue. Certains médicaments nécessitent une précision chirurgicale en raison de leur marge thérapeutique étroite. Il s'agit des médicaments à marge thérapeutique étroite (drogues où de petites variations de concentration sanguine peuvent entraîner une inefficacité ou une toxicité grave).
Pour des traitements comme la warfarine (anticoagulant) ou la levothyroxine (hormone thyroïdienne), les autorités réglementaires imposent souvent des limites plus strictes, typiquement comprises entre 90 % et 111 %. Ici, une variation de 20 % serait dangereuse. Inversement, pour les médicaments très variables (dont l'absorption fluctue naturellement beaucoup d'un individu à l'autre), des approches alternatives comme la bioéquivalence moyenne ajustée (SABE) peuvent élargir temporairement les limites acceptables, parfois jusqu'à 69,84-143,19 % pour le Cmax, sous conditions très spécifiques.
Impact clinique et perception des professionnels
Malgré la robustesse scientifique de la règle, des inquiétudes subsistent parmi certains praticiens. Une enquête de 2022 auprès de neurologistes a révélé que 28 % d'entre eux signalaient des problèmes occasionnels lors du changement de génériques d'antiépileptiques. Cependant, seuls 4 % attribuaient ces effets à la bioéquivalence elle-même. La plupart des cas étaient liés à des différences de forme galénique ou à l'effet Nocebo (où l'attente d'un effet négatif crée cet effet).
Les données de surveillance post-commercialisation parlent d'elles-mêmes. Sur plus de 2 000 génériques approuvés par la FDA entre 2003 et 2016, seulement 0,34 % ont nécessité des modifications d'étiquetage liées à des problèmes de bioéquivalence après leur mise sur le marché. Cela démontre que, dans la pratique, la règle des 80-125 % réussit à filtrer efficacement les produits qui poseraient un risque réel pour la santé publique.

Pourquoi cette harmonisation mondiale est cruciale
L'uniformité de ce standard est un atout majeur pour l'industrie pharmaceutique et les systèmes de santé. Que vous soyez traité aux États-Unis, en Europe via l'EMA, ou ailleurs, les critères sont quasi identiques. Cette harmonisation, poussée par le Conseil international d'harmonisation (ICH), permet aux laboratoires de développer un générique une seule fois pour le vendre dans plusieurs pays.
Cette efficacité réduit considérablement les coûts de développement. Un générique prend environ 18 à 24 mois à mettre au point, contre plusieurs années pour un nouveau médicament innovant. En abaissant les barrières à l'entrée, la règle des 80-125 % contribue directement à la baisse des prix des médicaments, rendant les soins accessibles à une population plus large. Aujourd'hui, les génériques représentent 90 % des ordonnances remplies aux États-Unis, tout en ne consommant que 23 % des dépenses totales en médicaments.
Avenir et évolutions technologiques
La science ne stagne jamais. Alors que la règle des 80-125 % reste solide pour la majorité des comprimés simples, elle fait face à des défis avec les formes complexes : inhalateurs, patchs cutanés ou gels ophtalmiques. Pour ces produits, la corrélation entre l'absorption systémique et l'effet local est plus difficile à modéliser.
Les agences réglementaires travaillent actuellement sur des approches basées sur la modélisation informatique (model-informed bioequivalence). L'objectif est d'utiliser des simulations physiologiques avancées pour prédire la bioéquivalence sans toujours recourir à des études humaines massives. De plus, la pharmacogénomique pourrait, d'ici 2030, amener à personnaliser ces critères selon le profil génétique du patient, ouvrant la voie à une médecine encore plus précise.
Est-ce que tous les génériques doivent respecter la règle des 80-125 % ?
Non. La règle s'applique à la majorité des médicaments oraux simples. Cependant, les médicaments à marge thérapeutique étroite (comme la warfarine) exigent des limites plus strictes, souvent entre 90 % et 111 %. À l'inverse, les médicaments très variables peuvent bénéficier de critères adaptés (SABE) qui élargissent légèrement les marges acceptables.
Pourquoi utilise-t-on un intervalle de confiance à 90 % et non 95 % ?
L'intervalle à 90 % est utilisé parce qu'il s'agit d'un test bilatéral. On alloue 5 % de risque d'erreur à la limite inférieure et 5 % à la limite supérieure, ce qui donne un risque global de 10 %. Cela permet de garantir que le produit n'est ni significativement moins efficace, ni significativement plus fort que la référence.
La règle des 80-125 % signifie-t-elle que mon générique contient moins de médicament ?
Absolument pas. Cette règle concerne l'absorption par le corps (biodisponibilité), pas la quantité de substance dans le comprimé. Tous les médicaments, génériques ou de marque, doivent contenir entre 95 % et 105 % de la dose indiquée sur l'emballage. La variation de 80-125 % porte sur la vitesse et l'étendue de l'absorption dans le sang.
Quels sont les paramètres AUC et Cmax ?
L'AUC (Surface sous la courbe) mesure l'exposition totale du corps au médicament au fil du temps. Le Cmax (Concentration maximale) mesure le pic de concentration atteint dans le sang et reflète la vitesse d'absorption. Pour être considéré bioéquivalent, un générique doit avoir ses valeurs d'AUC et de Cmax dont l'intervalle de confiance tombe dans la fourchette 80-125 % par rapport au médicament de référence.
Pourquoi transforme-t-on les données en logarithmes ?
Les concentrations de médicaments dans le sang ne suivent pas une distribution normale symétrique. La transformation logarithmique rend les données symétriques autour de zéro, permettant une analyse statistique valide et équitable des écarts positifs et négatifs. Sans cette étape, les calculs de bioéquivalence seraient biaisés.